
Medicine’s Dead Time
Somewhere, a patient with newly diagnosed mantle cell lymphoma is not waiting for a miracle. He is waiting for a system to finish knowing what it may already know.
His disease is not theoretical. Mantle cell lymphoma is a rare and typically aggressive form of non-Hodgkin lymphoma, accounting for roughly 6 percent of U.S. non-Hodgkin lymphoma cases, according to the Lymphoma Research Foundation (Lymphoma Research Foundation). It is the kind of diagnosis that turns time into an adversary. The calendar is no longer administrative furniture. It is biology. It is tumor burden, immune exhaustion, organ function, scan intervals, family logistics, and the dull, obscene arithmetic of whether the next option arrives before the body stops being able to use it.
He is not asking medicine to be careless. He is not demanding that a regulator bless every molecule with a good story and a billionaire sponsor. He is asking something simpler, and more difficult: if a therapy is moving through the system, if early signals are accumulating, if safety can be watched in something closer to real time, then what exactly is he waiting for?
That question used to sound impatient. Increasingly, it sounds scientific.
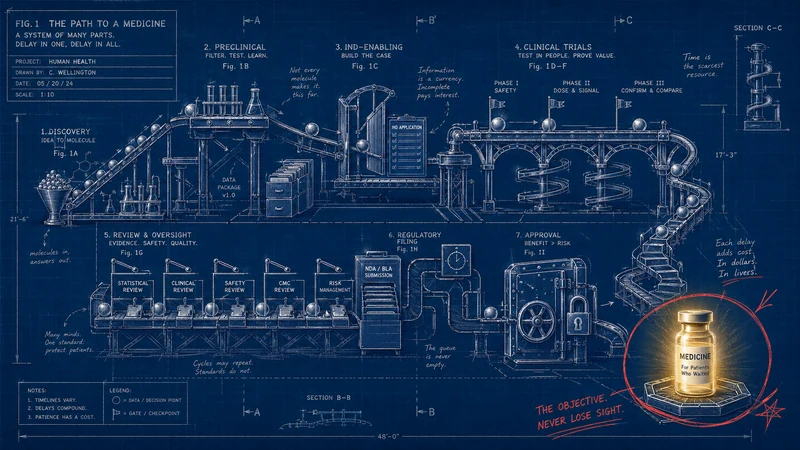
In April 2026, the U.S. Food and Drug Administration announced what, on paper, looked like a process reform: two proof-of-concept real-time clinical trials, one from AstraZeneca and one from Amgen. AstraZeneca’s TRAVERSE study is a Phase 2 multi-site trial in treatment-naïve mantle cell lymphoma, with participation from MD Anderson and the University of Pennsylvania. Amgen’s STREAM-SCLC trial is a Phase 1b study in limited-stage small cell lung carcinoma (FDA).
The agency described the move in the language of endpoints, safety signals, trial phases, and decision-making. Reuters sharpened the point: the program is meant to reduce “dead time” in drug development, with FDA Commissioner Marty Makary saying administrative tasks and paperwork consume about 45 percent of the time from early testing to submission for approval (Reuters).
Dead time. There it was, tucked inside the machinery. Not toxicity. Not disease progression. Not a failed endpoint. Dead time.
The phrase deserves to be taken literally.
The Quiet Scandal of Latency
For generations, modern medicine has organized its moral imagination around one great fear: the patient harmed by an intervention released too soon. This fear is not foolish. It is one of the reasons modern drug regulation exists. Bad drugs injure people. Weak evidence misleads doctors. False hope is not harmless simply because it arrives wearing a lab coat.
But the moral ledger has always had another side. The patient harmed by a bad therapy is visible. The patient who dies before a therapy arrives is usually not. One becomes an adverse event. The other becomes background mortality.
That asymmetry has shaped the emotional life of medicine. Action feels morally charged. Delay feels prudent. Approval feels like a decision. Waiting feels like the absence of one. Yet for the patient with aggressive cancer, delay is not the absence of harm. It is one of harm’s quieter forms.
This is the central ethical reversal hiding inside the FDA’s real-time trial initiative. The agency deserves credit for making dead time visible. Once visible, it can no longer be treated as mere procedure. The days, weeks, and months consumed by data cleaning, package preparation, sequential review, duplicative queries, fragmented systems, and national silos are not just clerical residue. In lethal disease, they are part of the clinical environment.
The old shorthand, “first do no harm,” was never quite as simple as its bumper-sticker form. The Hippocratic tradition does not command paralysis. It asks physicians not to intentionally injure. But modern medical culture has often converted that deeper ethic into a bias toward delay, as though the primary danger lies in doing something, while waiting occupies a clean moral space.
It does not. Waiting acts on patients. It acts through disease.
What the FDA Just Admitted
The most interesting thing about the FDA’s real-time trials announcement is not that it involves AI or that it sounds modern. Every institution now knows how to sprinkle AI over a press release like parsley on a hotel omelet. The more important admission is institutional: some delay is not necessary caution. Some delay is latency.
In the real-time clinical trials model, regulators can see aggregated safety and efficacy signals as studies progress, rather than waiting for companies to compile traditional submissions after the fact. Reuters reported that the FDA would not receive raw patient data, but aggregated information such as adverse-effect rates and tumor responses (Reuters). That matters. It means the reform is not a fantasy of total surveillance or regulatory impulsiveness. It is an attempt to move the evidentiary process from batch mode toward live mode.
The distinction is crucial. A conventional trial produces evidence, but the system often handles that evidence in slow pulses. Sites enroll patients. Sponsors collect data. Data are cleaned. Reports are assembled. Files are submitted. Regulators review. Questions come back. Responses go out. Meetings happen. Decisions emerge.
The disease does not wait for the paperwork to catch up.
Real-time trials do not abolish uncertainty. They do not make early signals definitive. They do not convert Phase 2 into omniscience. But they challenge an old assumption: that the pace of institutional review must lag far behind the pace of biological learning.
For the lymphoma patient outside the trial, this distinction is not procedural. It is existential. If a therapy looks promising, if safety signals are being watched, if regulators can see the shape of the evidence as it forms, the patient’s question becomes sharper: why must the system still behave as though knowledge only exists once it has been packaged?
The Lesson AIDS Activists Forced Into Medicine
Medicine has heard this kind of question before.
During the AIDS crisis, patients and activists forced regulators, researchers, and physicians to confront a brutal fact: traditional evidentiary timelines can become morally grotesque when people are dying quickly and no adequate treatments exist. The FDA’s accelerated approval pathway was established to allow earlier approval of drugs for serious conditions with unmet medical need based on surrogate endpoints reasonably likely to predict clinical benefit (FDA Accelerated Approval Program). A 2022 analysis in Therapeutic Innovation & Regulatory Science notes that accelerated approval was established by FDA regulation in 1992 in response to the AIDS epidemic (Therapeutic Innovation & Regulatory Science).
That history is often remembered as a triumph of activism over bureaucracy, and partly it was. But its deeper lesson is more uncomfortable. AIDS activists did not simply ask the system to move faster. They exposed an ethical accounting error. They insisted that the people dying outside the evidentiary process were part of the moral equation.
The lesson was not that every desperate patient should get every experimental drug. That would be a caricature, and a dangerous one. The lesson was that medicine cannot pretend that perfecting evidence has no human cost.
That distinction matters now because the tools are changing. AIDS activists had urgency. Today, medicine may finally be building instruments that make urgency safer.
From Slow Certainty to Continuous Learning
The future of drug development will not be shaped by one FDA pilot. It will be shaped by a larger scientific migration: from animal-heavy, sequential, paperwork-bound systems toward more human-relevant, computational, monitored, adaptive evidence systems.
The FDA’s own work on New Approach Methodologies points in this direction. The agency has described a shift toward human-relevant methods such as AI-powered models, organ-on-chip systems, and in silico modeling. It also notes a stark fact: more than 90 percent of drugs that appear safe in animals fail in humans (FDA New Approach Methodologies). That number should land like a hammer. It does not mean animal studies are useless. It does mean the old pipeline, for all its ritual authority, is full of uncertainty masquerading as prudence.
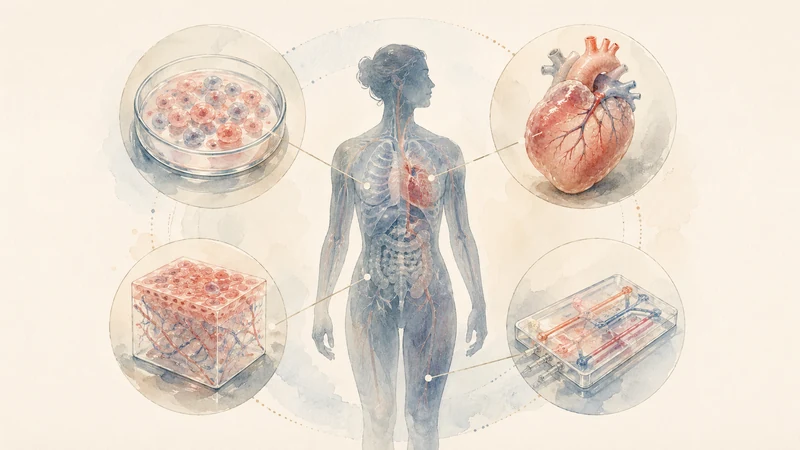
AI will not magically solve this. Anyone who says otherwise should be kept at least 500 yards from a clinical protocol. Models can overfit. Biological systems surprise. Organoids are not people. An organ-on-chip is not a marriage of liver, immune system, microbiome, age, sex, comorbidity, environment, and bad luck. Human beings remain defiantly inconvenient.
Still, the direction of travel is clear. Better cellular models, organ-level systems, real-world data, adaptive designs, and real-time monitoring can make uncertainty more observable. They can reveal signals earlier. They can help identify toxicity sooner. They can make trial design more responsive. They can help regulators distinguish between necessary biological time and unnecessary administrative time.
That is the real transformation. Not speed for its own sake. Not the Silicon Valley fever dream of “move fast and cure things.” The point is better safety. Older safety waits, batches, reviews, and approves. Newer safety models, monitors, adapts, escalates, and, when the evidence fails, withdraws.
The answer to bad acceleration is not ritualized delay. It is better instrumentation.
The National Border Is a Medical Artifact
Once evidence becomes more continuous, another problem comes into view: national drug-approval systems begin to look scientifically and morally outdated.
This is especially obvious in rare and lethal diseases. The patients are distributed across countries. The science is global. The sponsors are global. The investigators are global. The conferences are global. The datasets should be global. But the pathways remain stubbornly fragmented, split across national regulators, local reimbursement bodies, site-specific approvals, incompatible data systems, duplicated paperwork, and uneven access.
For a disease like mantle cell lymphoma, fragmentation is not a technical annoyance. It is slower learning. Slower learning means slower confidence. Slower confidence means slower access. And slower access means some patients deteriorate or die before the system finishes reconciling itself.
The World Health Organization has already moved partway toward this terrain. Its 2025 Global Action Plan for Clinical Trial Ecosystem Strengthening calls for sustainable, efficient, inclusive clinical-trial ecosystems that generate high-quality evidence for policy and practice (WHO Global Action Plan). The WHO also describes the plan as a way to help countries and stakeholders reform clinical trial systems so they are fit for purpose, inclusive, and locally led (WHO Departmental Update).
That last phrase matters. A global evidence system must not become a polished new instrument for rich countries to extract data from poor ones. It must not mean one imperial super-regulator. It must not mean lowering standards in the name of urgency. The point is not centralization for its own sake. The point is interoperable learning: shared endpoints, trusted data pipelines, real-time safety signals, harmonized evidence standards, faster recruitment, and regulatory cooperation that reflects the biological fact that cancer does not carry a passport.
The FDA pilot is admirable. But the moral horizon is larger than the FDA. If we can build more continuous evidence systems inside one country, the next question is whether we can build them across countries. If we cannot, then national borders become part of the disease environment.
The Case Against Recklessness
There is an obvious objection to all of this, and it is a serious one. Faster can hurt people.
The recent history of drug approval is not a clean morality play in which bold patients and brilliant innovators are forever thwarted by timid bureaucrats. The world is messier. Desperate patients can be exploited. Early signals can fade. Surrogate endpoints can mislead. Companies can wrap commercial pressure in compassionate language. Regulators can approve therapies whose benefits remain uncertain, while the costs, clinical burdens, and risks arrive immediately.
Relyvrio for ALS is a useful warning. Amylyx began voluntarily discontinuing marketing authorizations in the U.S. and Canada after the Phase 3 PHOENIX trial failed to confirm benefit (Amylyx). Elevidys is an even sharper safety caution. In 2025, the FDA said it had received three reports of fatal acute liver failure following Sarepta AAVrh74 gene therapies, requested suspension of distribution for Elevidys, and placed related trials on hold (FDA Elevidys Safety Action).
These cases should not be waved away. They are the reason serious people should distrust cheap accelerationism. Speed without truth is not compassion. It is roulette with better branding.
But these failures do not vindicate slow medicine as such. They vindicate disciplined learning. They argue for stronger monitoring, clearer uncertainty, better post-market obligations, faster reversals, and less tolerance for evidentiary fog. If a therapy is approved earlier, the system must be able to watch harder. If a signal collapses, the system must be able to withdraw faster. If risk emerges, the system must be able to act without theatrical surprise.
The ethical future is not “approve first, ask questions later.” It is “learn earlier, monitor continuously, act faster, and reverse course decisively.”
That is a harder standard than the old one. It demands more of regulators, sponsors, clinicians, data systems, and patients. It also demands more honesty from the rest of us. We cannot continue to praise caution as though it were costless, then avert our eyes from the patient whose disease outruns the process.
First, Count the Waiting
Return to the patient with mantle cell lymphoma. The trial is moving. The model is updating. Sites are enrolling. Tumor responses are being tracked. Safety signals are being watched. Somewhere, a regulator may be seeing the evidence sooner than the old system allowed.
And still, the patient waits.
The question is not whether he should be handed an unproven therapy because his suffering is real. Suffering alone does not make a drug work. The question is whether medicine’s inherited protocols still match its emerging capabilities. When evidence can be modeled more deeply, monitored more continuously, and shared more quickly, delay takes on a different moral weight.
The FDA’s real-time clinical-trials proof-of-concept should be celebrated because it points to an uncomfortable truth. Some of what we call caution is science. Some of it is latency. The first is necessary. The second is harder and harder to defend.
Medicine’s deepest moral failure in the AI-biology era may not be moving too fast. It may be moving slowly out of habit, prestige, liability, paperwork, and institutional muscle memory, while insisting that the only harms worth counting are the ones caused by action.
Time is not empty. Time is biological. Time is moral.
The next version of “first do no harm” must have the courage to count the harm of waiting.
References
- FDA Announces Major Steps to Implement Real-Time Clinical Trials
- US FDA to monitor clinical trial data in real time in pilot program aimed at speeding approvals
- Mantle Cell Lymphoma
- FDA Accelerated Approval Program
- Analysis of FDA’s Accelerated Approval Program Performance
- FDA New Approach Methodologies
- WHO Global Action Plan for Clinical Trial Ecosystem Strengthening
- WHO releases global action plan to strengthen clinical trial ecosystems
- Amylyx announces formal intention to remove RELYVRIO/ALBRIOZA from the market
- FDA Requests Sarepta Therapeutics Suspend Distribution of ELEVIDYS